|
|
The Genesis of ALS
Investigating mitochondria and calcium regulation
 |
| Gavriel David, M.D., Ph.D., has been assisted by graduate student Khanh T. Nguyen
on ALS research. |
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig’s Disease, is a devastating condition that causes progressive paralysis and death due to the degeneration of motor neurons and their motor nerve terminals that, normally, cause the skeletal muscles to contract and allow movement.
Though there is
no cure or effective treatment for ALS, Gavriel David, M.D., Ph.D., research associate professor of physiology and biophysics, has been researching the cause, with a view to finding therapy for the 30,000 Americans with ALS and the 5,000 new cases diagnosed every year.
In research published in Proceedings of the National Academy of Sciences, David and a team of researchers worked with mouse models to look into the mechanisms that make motor nerve terminals vulnerable to ALS degeneration, advancing previous research that investigated the process by which motor neuron terminals handle calcium that enters during activity.
As it turns out, regulating the uptake of calcium into the mitochondria is a very important function of the nerve terminal. If calcium isn’t sequestered, then calcium levels become too high, causing the nerve terminal to die and muscle contraction to fail.
“In ALS we believe something
goes wrong, perhaps with the ability of the mitochondria doing their calcium sequestration job,” says David. “We wanted to see whether something goes wrong in this important process— specifically, not late in the disease process but very early. If we can catch this process early, before the cell body has died, then there is a chance of correcting the problem.”
Looking at healthy mice, the team found that the mitochondria work like a battery, successfully taking up calcium while maintaining voltage. In the sick mice (those expressing mutated genes for superoxide dismutase 1, known
to cause ALS in some humans), the ability of the mitochondria to maintain power and uptake and regulate calcium was compromised very early in the process, even while all other functions seemed to be operating normally. The researchers believe that over time, the diminishing power of the mitochondria leads to continuous inefficiency in calcium sequestration, eventually killing the terminal and leading to paralysis and death, which happened in the case of the sick mice.
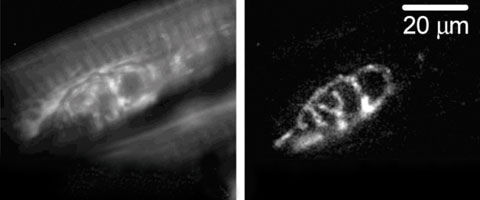 |
| The fluorescent dye rhodamine-123 is used to measure changes in the voltage of mitochondria, located inside motor nerve terminals. A motor nerve terminal from a mouse model of amyotrophic lateral sclerosis is first labeled with the dye, left, and then activated by electrical stimulation. Right, the dye “lights up” the regions in which the voltage decreased during stimulation. |
David is focusing his research
on what happens before paralysis and death. Believing the compromised regulation of calcium due to inefficiency in the mitochondria may
be a cause, he is testing whether agents that reduce calcium can help the mitochondria maintain efficiency.
Even this early research has potential for a therapy. “Possible treatment might involve agents that somehow reduce the amount of calcium that is absorbed, or agents that reduce the toxic effects of calcium after it is absorbed,” says David. “The tricky situation here
is you cannot eliminate the calcium completely or you’ll get paralysis.”
Several other Miller School scientists contributed to the research, which was supported by grants from the National Institutes of Health, Muscular Dystrophy Association, and Amyotrophic Lateral Sclerosis Association. The researchers are Khanh T. Nguyen, a graduate student in the neuroscience program; John Barrett, Ph.D., and Ellen Barrett, Ph.D., professors in the Department of Physiology and Biophysics; and Luis García-Chacón, M.D., Ph.D., now a resident in the Department of Pediatrics who contributed research as part of his graduate studies in the Department
of Physiology and Biophysics.
|
|
|